Plasma Cell Dyscrasias THE MERCK MANUAL, Sec. 11, Ch. 140,
Multiple Myeloma(Plasma Cell Myeloma; Myelomatosis)
A progressive neoplastic disease characterized by marrow plasmacytomas (plasma cell tumors) and overproduction of an intact monoclonal immunoglobulin (IgG, IgA, IgD, or IgE) or Bence Jones protein (free monoclonal or light chains).
Multiple myeloma is often associated with multiple osteolytic lesions, hypercalcemia, anemia, renal damage, and increased susceptibility to bacterial infections; production of normal immunoglobulin is impaired. The incidence is estimated at 2 to 3/100,000 persons, the male:female ratio is 1.6:1, and most patients are > 40 yr. The prevalence in blacks is twice that in whites.
Etiology and Pathogenesis of Plasma Cell Dyscrasias
The etiology is unknown. A relationship is suggested by finding Kaposi’s sarcoma-associated herpes virus in the dendritic cells cultured from myeloma patients. This virus encodes an interleukin-6 homologue; human interleukin-6 promotes myeloma growth and stimulates resorption of bone.
The specific cell of origin is unknown. Analysis of immunoglobulin gene sequences and cell surface markers suggests malignant transformation of a post-germinal center cell.
Pathology of Plasma Cell Dyscrasias
Diffuse osteoporosis or discrete osteolytic lesions develop, usually in the pelvis, spine, ribs, and skull. Lesions are due to bone replacement by expanding plasmacytomas or a factor secreted by malignant plasma cells (osteoclast-activating factor). The osteolytic lesions are usually multiple but occasionally are solitary intramedullary masses. Extraosseous plasmacytomas are unusual but may occur in any organ, especially the upper respiratory tract.
Plasmacytomas produce IgG in about 55% of myeloma patients and IgA in about 20%; of these IgG and IgA patients, 40% also have Bence Jones proteinuria. Light chain myeloma is found in 15 to 20% of patients; their plasma cells secrete only free monoclonal light chains ( or Bence Jones protein), and an M spike is usually absent on serum electrophoresis. Patients with the light chain subgroup tend to have a higher incidence of lytic bone lesions, hypercalcemia, renal failure, and amyloidosis than do other myeloma patients. IgD myeloma accounts for about 1% of cases; serum levels are often relatively low, and marked Bence Jones proteinuria (80 to 90% type ) is characteristic. Only a few cases of IgE myeloma have been reported. Nonsecretory myeloma (no identifiable M component in serum or urine) is very rare (
Amyloid deposits (see Ch. 18) occur in 10% of myeloma patients and are especially likely in those with Bence Jones proteinuria.
Persistent unexplained skeletal pain (especially in the back or thorax), renal failure, or recurrent bacterial infections are the most common presentations. Pathologic fractures and vertebral collapse are common; the latter may lead to spinal cord compression and paraplegia. Renal failure (myeloma kidney) may be caused by extensive cast formation in the renal tubules, atrophy of tubular epithelial cells, and interstitial fibrosis. Anemia, sometimes with weakness and fatigue, predominates in some patients, and a few have manifestations of the hyperviscosity syndrome (see under Macroglobulinemia, above). Lymphadenopathy and hepatosplenomegaly are unusual.
In the patient with a serum M protein, any one of three additional findings fulfills criteria for the diagnosis of myeloma: sheets or clusters of marrow plasma cells, osteolytic lesions (without evidence of metastatic carcinoma or granulomatous disease), or Bence Jones proteinuria > 300 mg/24 h.
Blood tests show a normocytic normochromic anemia with rouleaux formation. The WBC and platelet counts usually are normal. The ESR is often markedly elevated, sometimes > 100 mm/h, and BUN, serum creatinine, and serum uric acid are frequently elevated. A low anion gap is sometimes present. Hypercalcemia is found at diagnosis in about 10% of patients. The serum level of 2-microglobulin is frequently elevated and correlates with myeloma cell mass.
Proteinuria is common because of excess synthesis and secretion of free monoclonal light chains. Chemical paper strip tests of urine do not reliably detect Bence Jones protein, and the heat test is often misleading, but sulfosalicylic acid and toluene sulfonic acid are useful screening tests. Significant albuminuria is rare in myeloma; its presence suggests coexisting amyloidosis or light chain deposition disease.
Serum protein electrophoresis shows a tall, narrow, homogeneous M spike in about 80% of patients; the mobility of the M spike may lie anywhere from the 2 to the slow region. The remaining 20% of patients synthesize only free monoclonal light chains (Bence Jones protein), and their serum electrophoretic patterns display hypogammaglobulinemia without an M spike. However, in essentially all patients with light chain myeloma, a homogeneous M spike is demonstrable on protein electrophoresis of concentrated urine. Immunoelectrophoresis or immunofixation using monospecific antisera identifies the immunoglobulin class of the M spike in serum or urine.
X-rays of the bones may show typical punched-out lytic lesions or diffuse osteoporosis. Osteoblastic lesions are rare, and thus radionuclide bone scans usually are not helpful. MRI may be useful, particularly in predicting outcome in patients with early-stage disease.
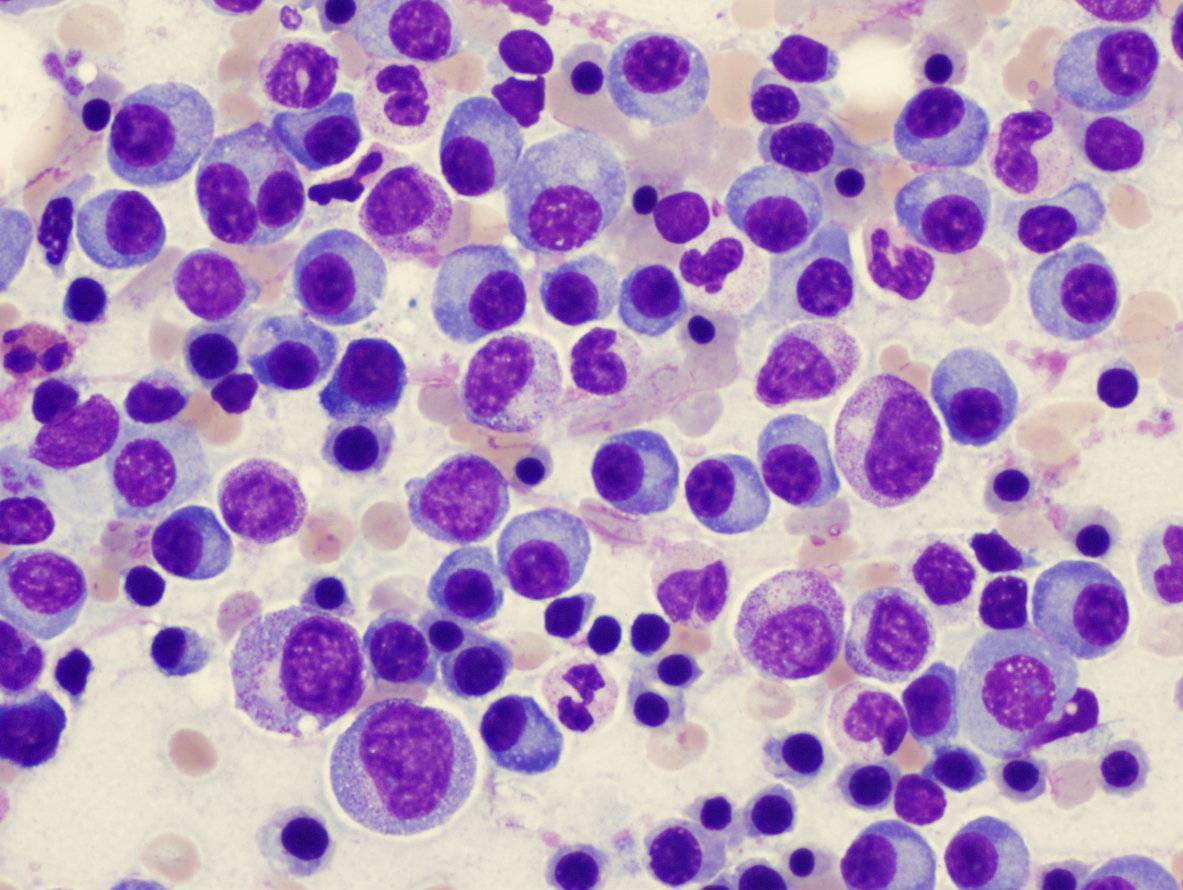
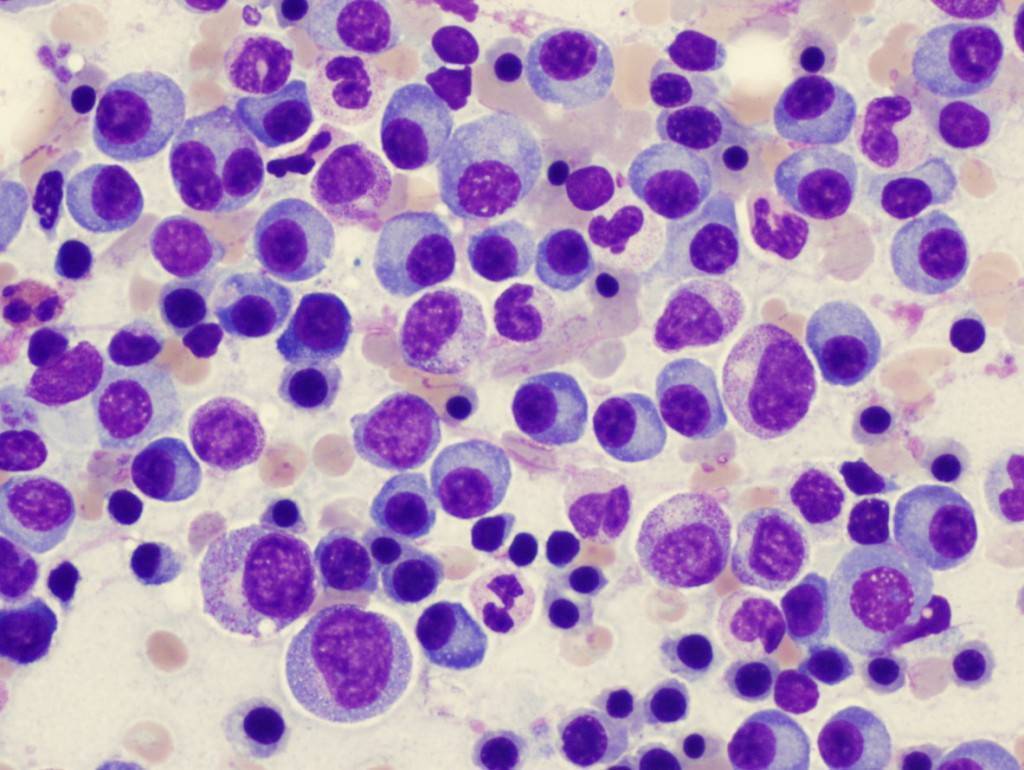
Bone marrow aspiration and biopsy usually show increased numbers of plasma cells at various stages of maturation; rarely is the number of plasma cells normal. Plasma cell morphology does not correlate with the class of immunoglobulin synthesized. Although sheets and clusters of plasma cells are diagnostic of marrow tumors, myeloma is a patchy disease, and often only modest nonspecific plasmacytosis is observed initially.
The disease is progressive, but good management improves the quality and duration of life. About 60% of patients treated show objective improvement. Median survival is about 2.5 to 3 yr, but this varies according to the extent of disease at diagnosis, adequacy of supportive measures, and response to drugs. At diagnosis, high levels of M protein in serum or urine, elevated serum 2-microglobulin levels, diffuse bone lesions, hypercalcemia, anemia, and renal failure are unfavorable prognostic signs.
Maintenance of ambulation is vital for protection from hypercalcemia and bone quality. Analgesics and palliative doses of radiotherapy (18 to 24 Gy) administered to localized areas of symptomatic bone involvement relieve pain significantly. However, radiotherapy may impair the patient’s ability to receive cytotoxic doses of systemic chemotherapy. All patients should receive pamidronate (90 mg/mo IV), which reduces skeletal complications and lessens bone pain and the need for analgesics. This treatment may also improve survival.
Adequate hydration is essential. (Dehydration before intravenous dye load may precipitate acute oliguric renal failure in patients with Bence Jones proteinuria.) Even patients with prolonged massive Bence Jones proteinuria (>= 10 to 30 g/day) may have little evidence of impaired renal function if they are well hydrated (urine output > 2000 mL/day).
Prednisone 60 to 80 mg/day po is useful for controlling hypercalcemia; pamidronate may be helpful in refractory cases (see above). Although most patients do not require allopurinol, a dose of 300 mg/day po controls hyperuricemia. Antibiotics are indicated for documented bacterial infection, but prophylactic use is not recommended. Most patients experience infections only during chemotherapy-induced neutropenia. Prophylactic IV immunoglobulin has been shown in some studies to reduce the risk of infections. However, it should be reserved for select patients with recurrent infections. Transfusion of packed RBCs is indicated for symptomatic anemia. Recombinant erythropoietin is very effective in reversing the anemia, especially in patients with renal dysfunction; however, its use should be limited to patients for whom chemotherapy does not raise Hb.
Chemotherapy: Response to chemotherapy is indicated by decreases in serum or urine M protein. Conventional chemotherapy rarely eliminates M protein; however, objective improvement (a >= 50% reduction in serum or urine M protein) often follows use of oral alkylating drugs (melphalan or cyclophosphamide). Median survival may be extended three- to sevenfold.
Prednisone (1 mg/kg/day for 4 days q 4 to 6 wk) or another glucocorticosteroid should be used with melphalan or cyclophosphamide. Glucocorticosteroids may be used alone to treat patients with newly diagnosed myeloma.
Melphalan may be given intermittently (0.25 mg/kg/day for 4 days q 4 to 6 wk). About 2 wk after administration, a WBC count must be obtained at nadir; if WBCs are > 3000/µL, the dose may be inadequate. Prednisone given intermittently (1 mg/kg/day for 4 days q 6 wk) may improve response to melphalan. Cyclophosphamide (200 mg/day for 5 to 7 days, then 50 to 100 mg/day for maintenance) appears to be as effective as melphalan. Because leukopenia and thrombocytopenia may develop with use of these drugs, WBC and platelet counts must be closely monitored.
Acute nonlymphocytic leukemia or myelodysplasia follows in a minority of responding patients and is likely related to length of exposure to mutagenic agents (alkylating drugs, irradiation). Thus, care must be taken to ensure that patients receive therapy for the shortest duration needed. Continuation of chemotherapy beyond this point has not been shown to improve survival.
High-dose therapy, ie, the use of more aggressive multidrug regimens requiring hematopoietic support, appears promising, although improvement in overall survival has been difficult to demonstrate in randomized trials. Because alkylating drugs should be avoided in high-dose therapy (they damage hematopoietic stem cells), infusional vincristine and doxorubicin with oral dexamethasone should be considered before transplantation. High-dose therapy followed by autologous bone marrow transplant in patients who previously received several courses of conventional chemotherapy was shown to improve remission rates and survival in one study.
Autologous peripheral stem cell support has largely replaced bone marrow transplantation for myeloma patients undergoing myeloablative chemotherapy. This procedure should be considered in patients
Maintenance therapy has been tried with nonchemotherapeutic drugs, including interferon, which prolongs remission but has little effect on overall survival. Glucocorticosteroids are being evaluated.
Leave a Reply
You must be logged in to post a comment.